paired end sequencing read length
The Illumina paired-end sequencing technology can generate reads from both ends of target DNA fragments which can subsequently be merged to increase the overall read length. 96 Gbp 105 Gbp 091 lanes àround up to 1 lane because we do not sell partial lanes.

Schematic Representation Of The Random Amplified Polymorphic Dna Rapd Download Scientific Diagrams Sequence Analysis Segmentation Dna Sequence
These fragments are.
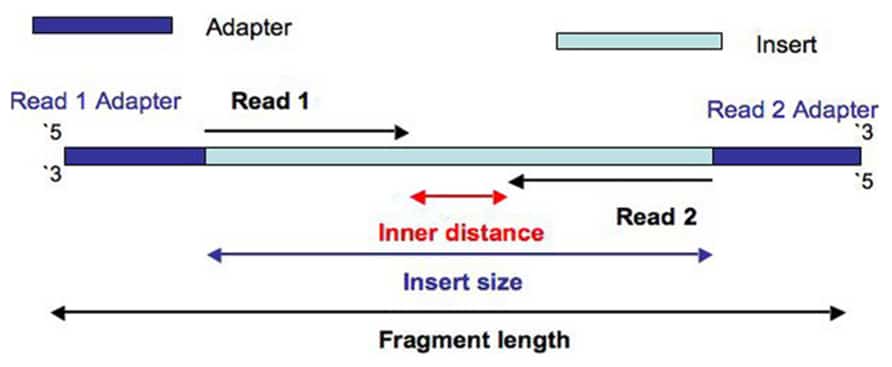
. If assembling the reads into the reconstructed DNA sequence is like doing a puzzle long reads equate to larger puzzle pieces. The current read length that is standard for many experiments is paired-end 100 bp reads and there is also the possibility of running paired-end 300 bp reads. First mate ends in the second mate.
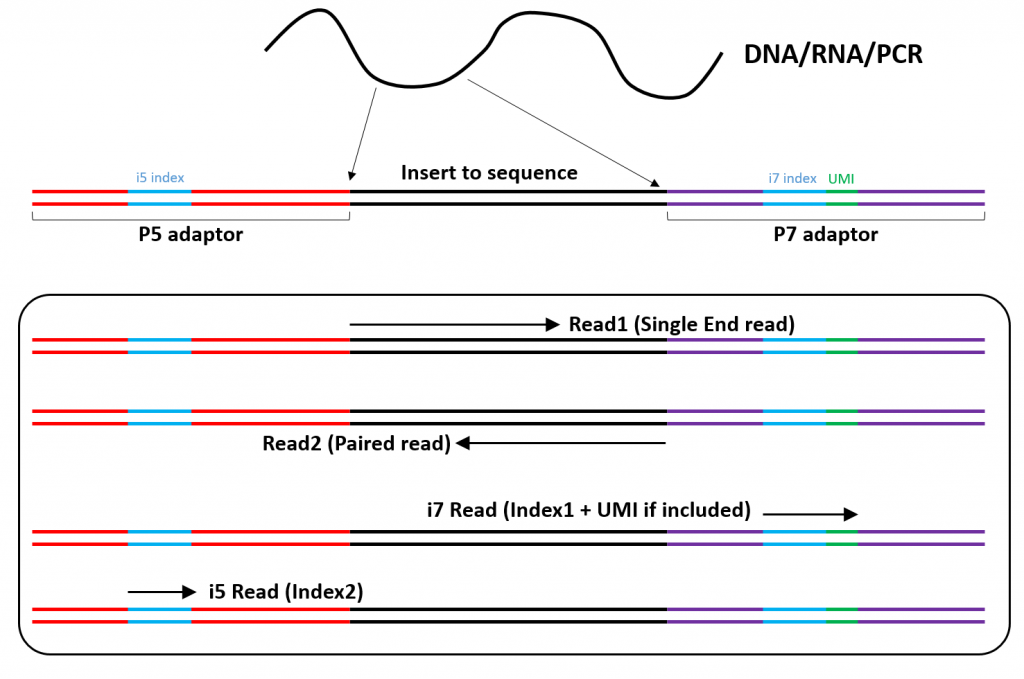
Next-generation sequencing NGS read length refers to the number of base pairs bp sequenced from a DNA fragment. Combinatorial dual CD indexes have eight unique dual pairs of index adapters so most libraries share sequences on the i7 or i5 end. The number of cycles is selected on the sequencing machine before.
Small RNA Analysis Due to the short length of small RNA a single read usually a 50 bp read typically covers the entire sequence. Sequencing read lengths correspond directly to the sequencing reagents used on an NGS. After sequencing the regions of overlap between reads are used to assemble and align the reads to a reference genome reconstructing the full DNA sequence.
No overlap the sequences do not overlap. Longer reads can provide more reliable information about the relative locations of specific base pairs. First mate begins in the second mate in this case the primeradapters from the other ends are sequences a scenario referred.
Read length describes the average length of the sequencing reads produced ie the number of base pairs sequenced and is sequencing-platform specific. Paired-end DNA sequencing reads provide high-quality alignment across DNA regions containing repetitive sequences and produce long contigs for de novo sequencing by filling gaps in the consensus sequence. Of reads x read length 1000000000 350000000 x 300 1000000000 105 Gbp Calculate desired Gbp.
MO 2 x 151bp. Paired end sequencing refers to the fact that the fragment s sequenced were sequenced from both ends and not just the one as was true for first generation sequencing. HO 2 x 151bp.
Modern nextgeneration sequencing platforms offer a range of read configurations such as single-read SR and paired-end PE sequencing with 75 bp per read 100 bp per read and 150 bp per read as routinely used methods. For Illumina kits for example you include R1 and R2 length in the sample sheet. Many platforms Illumina Genome Analyzer Applied Biosystems SOLID Helicos HeliScope are currently able to produce ultra-short paired reads of lengths starting at.
Jul 24 2019 at 1819. The current read length that is standard for many experiments is paired-end 100 bp reads and there is also the possibility of running paired-end 300 bp reads. Paired-end reads are required to get information from both 5 and 3 ends of RNA species with stranded RNA-Seq library preparation kits.
The length of the sequence reads then is determined by the number of sequencing cycles. Next-generation sequencing NGS read length refers to the number of base pairs bp sequenced from a DNA fragment. Jul 25 2019 at 2021.
MO 2 x 151bp. Unique dual UD indexes have distinct unrelated index adapters for both index reads. Maximum Read Length.
We show that the fragment length is a major driver of. Index adapter sequences are eight or 10 bases long. A read length of 50 bp sequences most small RNAs plus enough of the.
For example one read might consist of 50 base pairs 100 base pairs or more. 12 Gbpx 80 96 Gbp Calculate of lanes required. In addition to producing twice the number of reads for the same time and effort in library preparation sequences aligned as read pairs enable more accurate read alignment and the ability to detect insertion-deletion indel.
For a 150 cycle kit you can perfectly run 75-75 125-25 and even 50-10. More recently the lengths of reads have increased substantially and sequencers have been improved to allow for the sequencing of both ends of a fragment to allow for paired-end sequences. After sequencing the regions of overlap between reads are used to assemble and align the reads to a reference genome reconstructing the full DNA sequence.
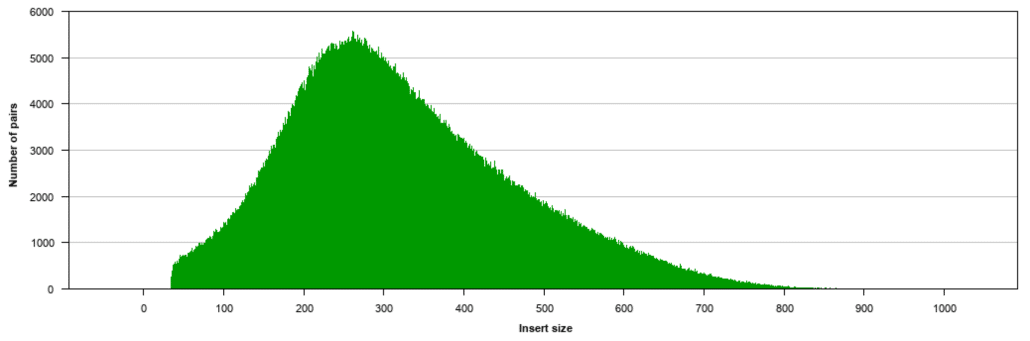
Genome Sequencing Example 1 Calculations Calculate expected Gbpper lane of HiSeq4000 PE150. A good choice for read length is closely tied to the insert size of the sequencing library ie how long the individual DNA fragments are that are sequenced. The explanation for this is that paired-end sequencing always starts at the endings of the fragment where the primer attaches creating read 1 and after a turnaround stage read 2 see Fig.
However some paired-end sequencing data show the presence of a subpopulation of reads where the second read R2 has lower average qualities. During sequencing it is possible to specify the number of base pairs that are read at a time. Sequencing read lengths correspond directly to the sequencing reagents used on an NGS.
Merging paired-end reads via novel empirically-derived models of sequencing errors BMC Bioinformatics. Next-generation sequencing technology is enabling massive production of high-quality paired-end reads. Depending on the length of your input and the length being sequences there are several ways your sequences in the mate files end up overlapping if there is any overlapping at all.
Paired-end DNA sequencing also detects common DNA rearrangements such as insertions deletions and inversions. The maximum distance x for a pair considered to be properly paired SAM flag 02 is calculated by solving equation Phix-musigmaxLp0 where mu is the mean sigma is the standard error of the insert size distribution L is the length of the genome p0 is prior of anomalous pair and Phi is the standard cumulative distribution function. Index adapter sequences are eight bases long.
HO 2 x 151bp. Paired-end sequencing facilitates detection of genomic rearrangements and repetitive sequence elements as well as gene fusions and novel transcripts. Since read lengths have increased substantially over recent years and will continue to increase we decided to determine whether longer reads are more beneficial for RNA-seq DEG and isoform determination.
As long as you dont exceed the maximum number of cycles you will be fine. There already exist tools for merging these paired-end reads when the target fragments are equally long.

Rna Extraction Method Read Length And Sequencing Layout Single End Versus Paired End Contribute Strongly To Var Interactive Notebooks Method Gene Expression

Diffusion Maps For High Dimensional Single Cell Analysis Of Differentiation Data Analysis Differentiation Cell
Illumina Short Read Sequencing Lausanne Genomic Technologies Facility

Deseq2 Moderated Estimation Of Fold Change And Dispersion For Rna Seq Data In Comparative High Throughput Sequencing Assa Molecular Biology Data Estimation
How Short Inserts Affect Sequencing Performance

The Teacher S Guide Is An Awesome Aggregate Site With Tons Of Resources For Resources Liste Interactive Learning Learning To Read Games Fiction And Nonfiction

Design Considerations Functional Genomics Ii
What Are Paired End Reads The Sequencing Center

Illumina Doubles Output Of Benchtop Sequencer Miseq To 15 Gb What Is Epigenetics Exome Sequencing Epigenetics Doubles
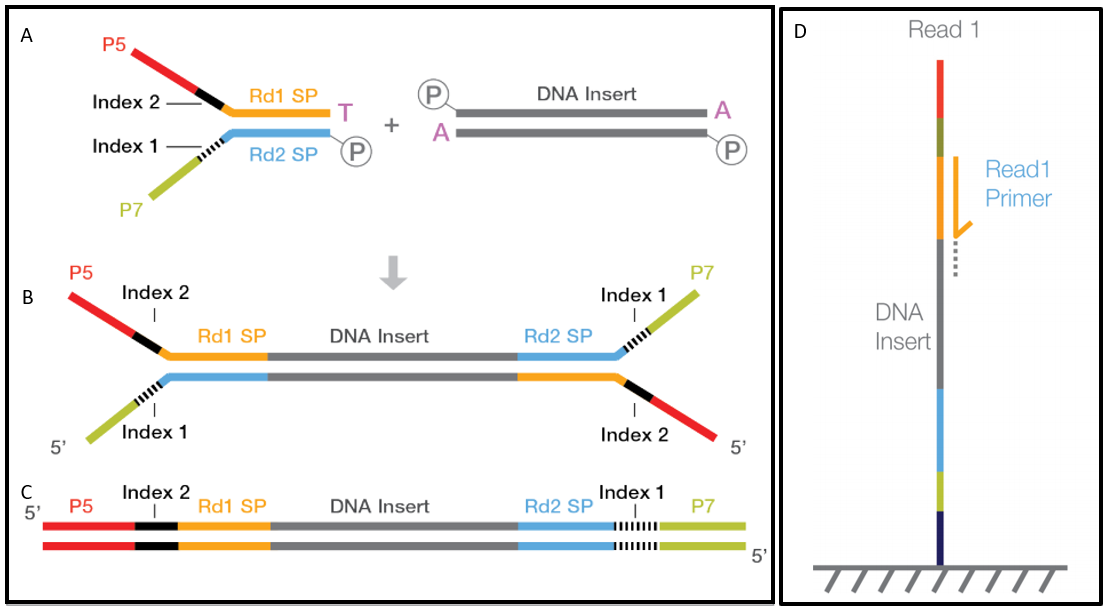
How Short Inserts Affect Sequencing Performance
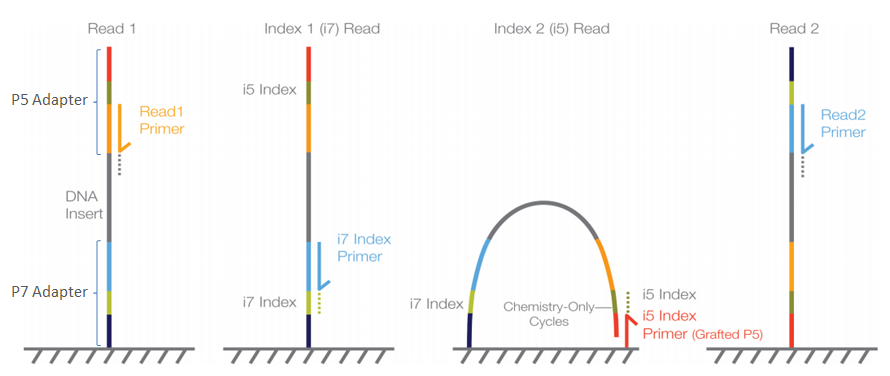
Adapter Trimming Why Are Adapter Sequences Trimmed From Only The 3a Ends Of Reads

A Team Led By Scientists At Bgi Tech Solutions Have Developed A New Method Soapfuse To Identify Fusion Transcripts From Algorithm Data Analysis How To Apply
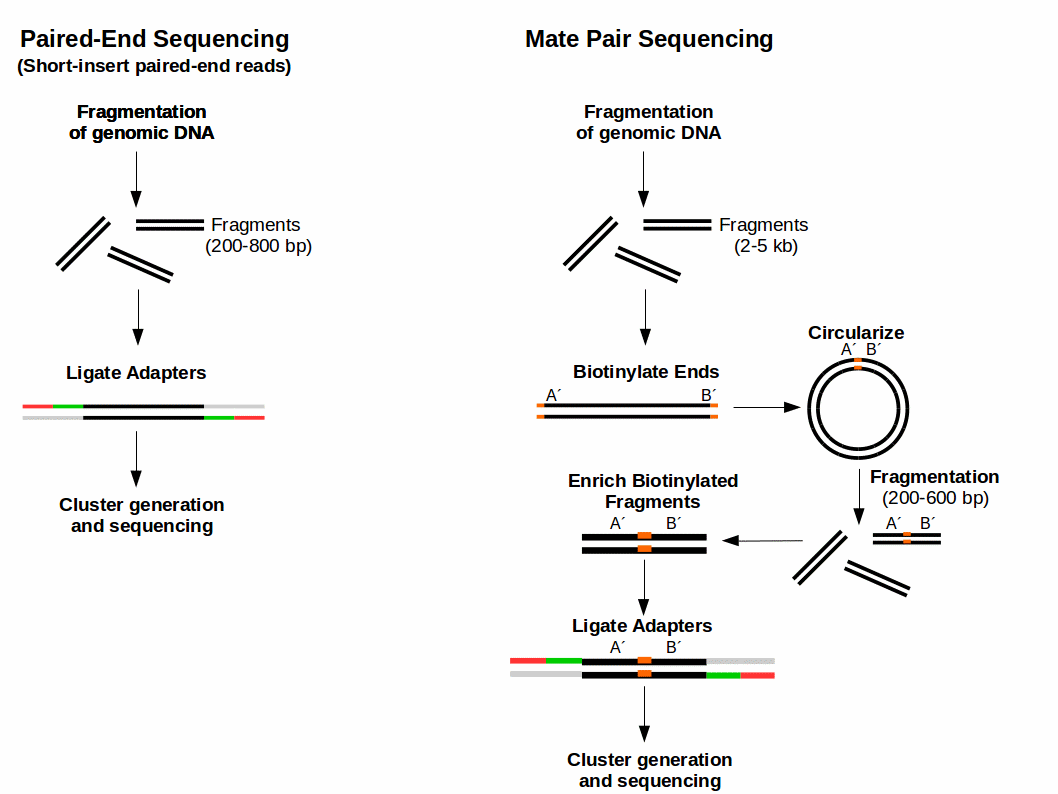
What Is Mate Pair Sequencing For
What Are Paired End Reads The Sequencing Center

Statquest Pca Clearly Explained Explained Statistical Analysis Informative

Funpat Function Based Pattern Analysis On Rna Seq Time Series Data Dynamic Expression Data Nowadays Obtained U Analysis Functional Analysis Rna Sequencing

Rna Extraction Method Read Length And Sequencing Layout Single End Versus Paired End Contribute Strongly To Var Interactive Notebooks Method Gene Expression

Deseq2 Moderated Estimation Of Fold Change And Dispersion For Rna Seq Data In Comparative High Throughput Sequencing Assa Molecular Biology Data Estimation

Pin On Statistical Analysis